

The Environment for Tree Exploration (ETE) is a Python programming toolkit that assists in the automated manipulation, analysis and visualization of phylogenetic trees (although clustering trees or any other tree-like data structure are supported).
ETE is currently developed as a tool for researchers working in phylogenetics and genomics. If you use ETE for a published work, please cite:
Jaime Huerta-Cepas, Joaquín Dopazo and Toni Gabaldón. ETE: a python Environment for Tree Exploration. BMC Bioinformatics 2010, 11:24.
- The official web site of ETE is at http://etetoolkit.org
- News and announcements are usually posted on twitter: http://twitter.com/etetoolkit
This branch of code refers to ETE v3.0, which is currently in BETA state. Latest stable release is 2.3 (repository in branch 2.3: https://github.com/jhcepas/ete/tree/2.3)
Note that ETE v3 has changed the name of the module from ete2 to ete3, so both versions can coexist. The main different between both is that the later is a Python 2.7/3.4 compatible.
ETE v3 support python 2.7+ and 3.4+
Newick (including several sub-types), Extended Newick / New Hampshire Extended (NHX), PhyloXML and NeXML
Trees are loaded as a succession of TreeNode objects connected in a hierarchical way. Each TreeNode instance contains methods to operate with it independently. This is, although the top-most Tree node instance represents the whole tree structure, any child node can be used independently as a subtree instance.
Available (per node) operations include:
- Iteration over descendant or leaf nodes.
- Tree traversing: post-order, pre-order, level-order-
- Search (descendant) nodes by their properties.
- Root / Unroot
- Calculate branch-length and topological distances among nodes.
- Node annotation (add custom features and properties to nodes)
- Automatic tree pruning
- Tree structure manipulation (add/remove parent, children, sister nodes, etc.).
- Newick and extended newick (including annotations) writing
- shortcuts and checks: "len(Node)", "for leaf in Node", "if node in Tree", etc.
- comparison and topology distances
ETE provides specific methods to load, analyze and manipulate phylogenetic results. Thus, a PhyloTree instance is provided, which extends the standard Tree functionality with phylogenetics related methods. Most notably:
- Link trees with Multiple Sequence Alignments (MSAs).
- Automatic detection of species codes within family gene-trees
- Node monophyly checks.
- Orthology and paralogy detection based on tree reconciliation or species overlap.
- Relative dating of speciation and duplication events.
- Combined visualization of trees and MSA.
- Duplication aware tree decomposition
ETE 2.3+ provides also a set of command line tools to perform common tasks. Most notably:
- ete build: allows to build phylogenetic tree using a using a number of predefined built-in gene-tree and species-tree workflows.
- ete mod: modify tree topologies directly from the command line. Allows rooting, sorting leaves, pruning and more
- ete annotate: add features to the tree nodes by combining newick and text files.
- ete view: visualize and generate tree images directly form the command line.
- ete compare: compare tree topologies based on any node's feature (i.e. name, species name, etc) using the Robinson-Foulds distance and edge compatibility scores, even for trees of different size.
- ete ncbiquery: query the ncbi taxonomy tree directly from the database.
- ete generate: generate random trees, mostly for teaching and testing
A programmatic tree rendering engine is fully integrated with the Tree objects. It allows to draw trees in both rectangular and circular modes. The aspect of nodes, branches and other tree items are fully configurable and can be dynamically controlled (this is, certain graphical properties of nodes can be linked to internal node values).
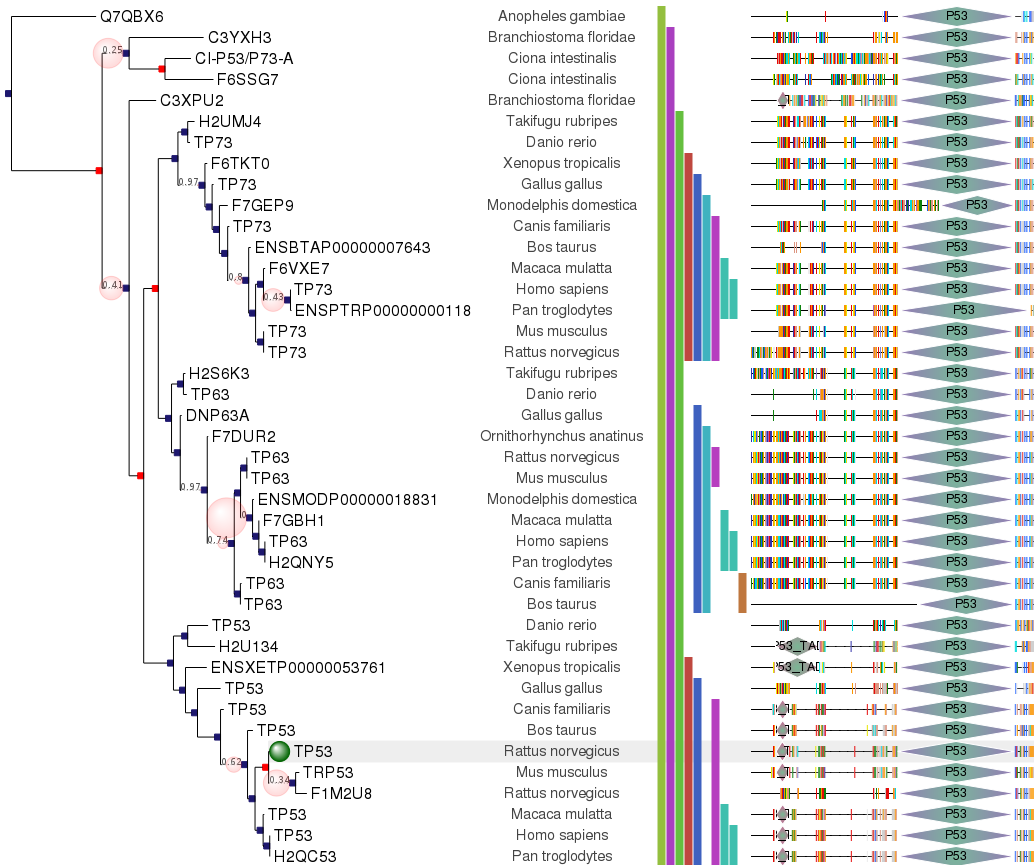
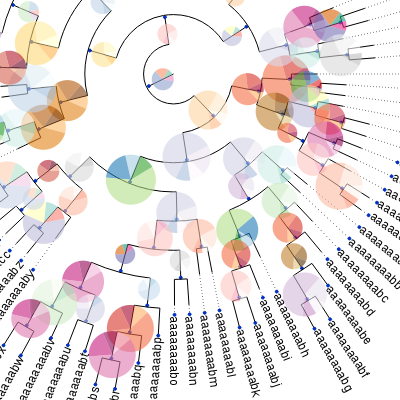
More examples at http://etetoolkit.org/gallery
Trees can also be visualized interactively using a built-in Graphical User Interface (GUI) or exported as PNG images or SVG/PDF vector graphics images.
The preferred way to report a problem or request/disccuss new features is by opening a new issue at http://github.com/jhcepas/ete/issues. (Please, make sure there is no other issues pointing to the same topic)
- There is a mailing list providing user technical support at https://groups.google.com/d/forum/etetoolkit . In order to avoid spam, messages from new users are moderated. Expect some delay until your first message and account is validated.
- For general questions on how to use ETE in bioinformatic projects, the BioStars community (http://biostars.org) provides an excellent and broader help desk. Please feel free to raise any question there and tag it with the "etetoolkit" label.
- Contributions to the main code, unittests and documentation are very
welcome. ETE's main source code is hosted at http://github.com/jhcepas/ete. Feel free to clone the repo and make pull requests.
- There is also a chat room for developers