Our decontaminer web server is under maintenance, so we are making it available here. Please read the decontaminer publication for more details.
https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-2684-x
Compressed folder "decontaMiner_1.4.tar.gz" contains latest version of the tool. Required databases can be downloaded from link mentioned below.
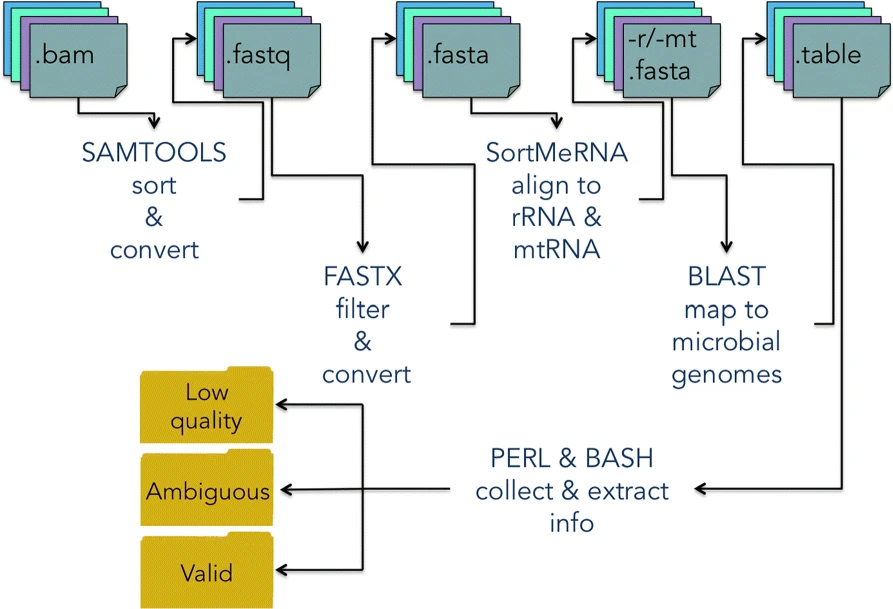
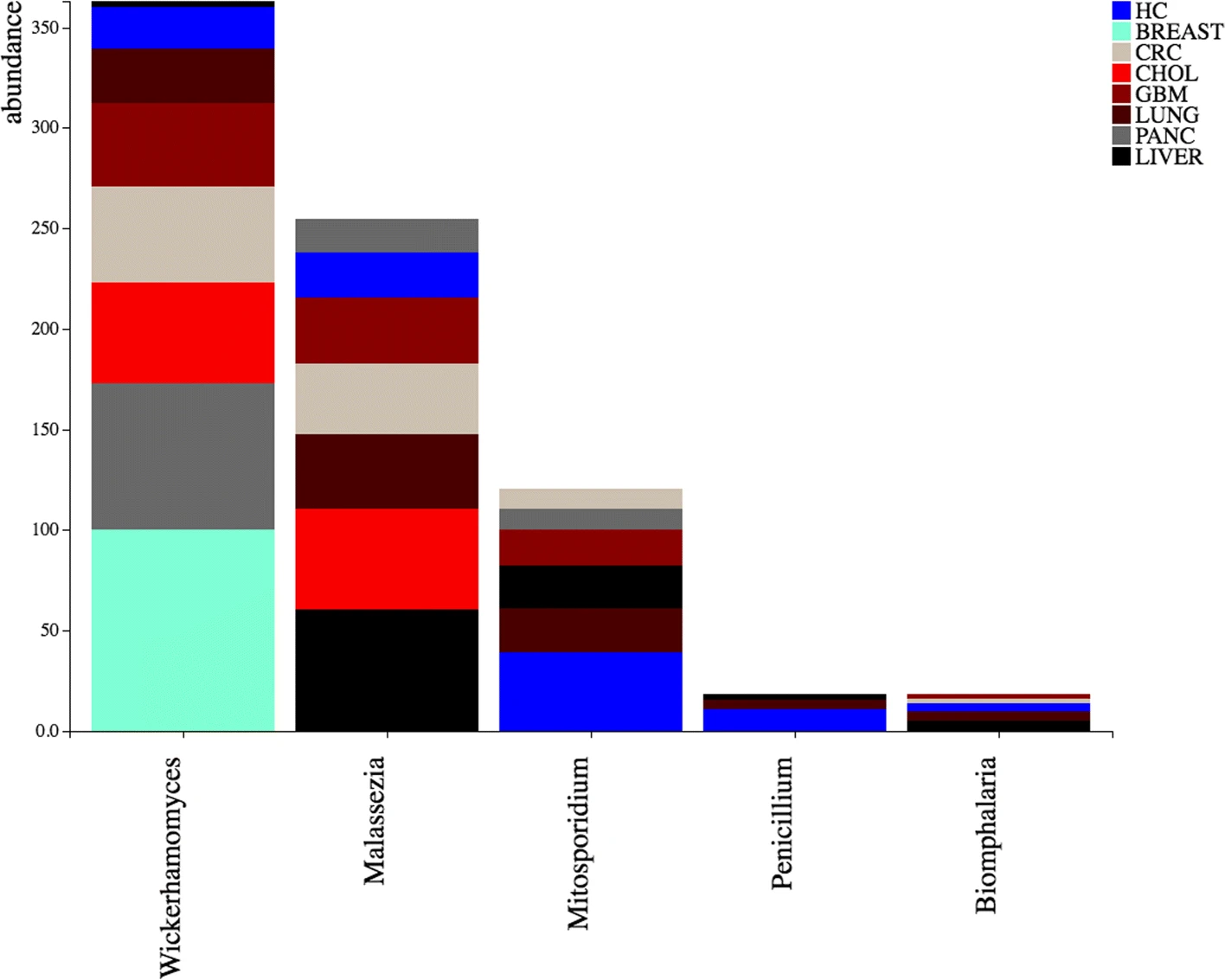
DecontaMiner, a tool to unravel the presence of contaminating sequences among the unmapped reads. It uses a subtraction approach to identify bacteria, fungi and viruses genome contamination. DecontaMiner generates several output files to track all the processed reads, and to provide a complete report of their characteristics. The good quality matches on microorganism genomes are counted and compared among samples. DecontaMiner builds an offline HTML page containing summary statistics and plots. The software is freely available at http://www-labgtp.na.icar.cnr.it/decontaminer.
https://drive.google.com/drive/u/2/folders/1UQCiuUVnS5TpkT0We2AkRVew-km_gR_u
Please find the code here https://rpubs.com/amarinder1/deontaminer or https://github.com/amarinderthind/decontaminer/tree/master/R_decontaminer_visualizationScript
if you have fasta and idx path as listed below
You should mention this in configuration file in the following way
RIBO_DB=DB/HUMAN_RNA
RIBO_NAME=rRNA
https://drive.google.com/drive/u/2/folders/1B9WNJc1cGY_LIi2XGwkQ0h_9916_A8Ij
Decontaminer expects the following Paired end reads format:
@A00121:137:HTLF3DSXX:3:1110:3097:35571/1
@A00121:137:HTLF3DSXX:3:1110:3097:35571/2
If you have other reads format (like below) format of PR reads, you can rename/change from
@A00121:137:HTLF3DSXX:3:1110:3097:35571 0:N: 00
@A00121:137:HTLF3DSXX:3:1110:3097:35571 1:N: 00
To following using simple linux command (mentioned below), with this command it will be converted to the required format like below:
@A00121:137:HTLF3DSXX:3:1110:3097:35571/1
@A00121:137:HTLF3DSXX:3:1110:3097:35571/2
Here is an example of Linux command
sed 's/ 0:N:0://1/g' inputfile > outputfile
sed 's/ 1:N:0://2/g' inputfile > outputfile